Quantitative Analyse von hochpolaren Pestiziden in Lebensmitteln mit SFC/MS
Da sich die Erzielung einer ausreichenden Retention und günstigen Trennung bei der normalen Chargenanalyse von hochpolaren Pestiziden aufgrund ihrer chemischen Eigenschaften als schwierig erwiesen hat, wird für die LC/MS/MS-Analyse eine Reihe von Einzelanalysemethoden eingesetzt. Um dieser Situation abzuhelfen, entwickelt EURL-SRM (Stuttgart, Deutschland), ein Mitglied der EU-Referenzlaboratorien, das für die Entwicklung individueller Analysemethoden zuständig ist, eine Chargenanalysemethode mit der Bezeichnung „QuPPe (Quick Polar Pesticides)“ für hochpolare Pestizide, die mit der Vorbehandlung mit der QuEChERS-Methode sowie mit normalen Chargenanalysemethoden schwer zu analysieren sind. Dieses Verfahren schlägt mehrere Methoden vor, um jeder Probe und jeder chemischen Zielverbindung gerecht zu werden (M. Anastassiades et al; QuPPe of EURL-SRM (Version 9.1; 2016)).
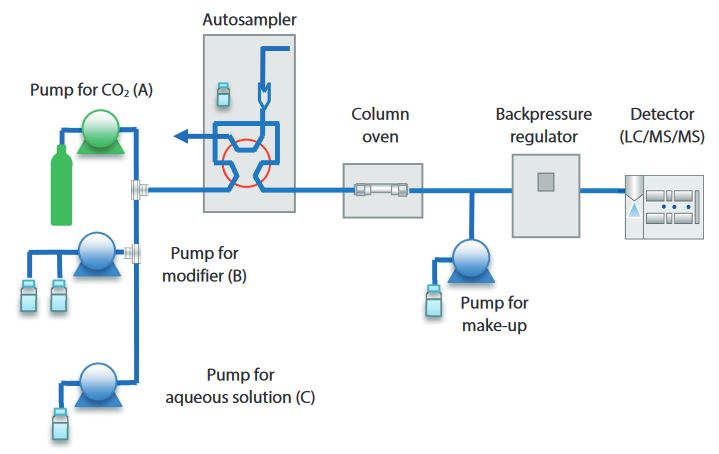
Bisher wurde bei der Analyse hoch polarer Pestizide mit LC/MS/MS eine Vielzahl von Trennmethoden verwendet, darunter HILIC-Modus, Mischmodus, Normalphase und Umkehrphase. Alle diese Methoden haben jedoch Einschränkungen hinsichtlich der chemischen Verbindungen, die zusammen analysiert werden können. Im Gegensatz hierzu bietet die überkritische Flüssigkeitschromatographie (SFC) den Vorteil, dass sie aufgrund der Eigenschaften der verwendeten mobilen Phase in der Lage ist, ein breites Spektrum chemischer Verbindungen auf einmal zu trennen. Da sich das Trennverhalten bei der SFC auch bei Verwendung einer Säule desselben Trennmodus von dem bei der LC unterscheidet, kann die SFC darüber hinaus für die Analyse von chemischen Verbindungen wirksam sein, bei denen Retention und Trennung bei LC schwierig sind. In diesem Artikel wird ein Beispiel für die Chargenanalyse von hochpolaren Pestiziden mit der SFC vorgestellt.
In diesem Experiment wurde untersucht, ob ein Zusatz einer geringen Menge an Wasser, zum für die SFC-Analyse üblicherweise verwendeten organischen Modifikator, die Elution und Trennung hochpolarer Pestizide ermöglicht. Um diese Untersuchung zu vereinfachen, wurde eine Niederdruckgradientenpumpe (LPGE) als Pumpe B verwendet, und der Modifikator wurde automatisch durch mobiles Phasenmischen vorbereitet.
Tabelle 1 SFC/MS-Analysebedingungen
| Supercritical Fluid Chromatography (SFC) | Massenspektrometrie (MS) | ||
|---|---|---|---|
| SFC | Nexera UC System | LC-MS/MS | LCMS-8060 |
| Analytische Säule | Restek Ultra Silica (150 × 2.1 mm 3 μm) | Ionisationsmodus | Beheizte ESI |
| Säulentemperatur | 50° C | Scan-Geschwindigkeit | 15,000 u/sec |
| Flussrate | 0.8 mL/min (0.6 mL/min 13-22 min) | MRM Dwell Time | 3 msec |
| Pumpe A | CO2 | Pause | 1 msec |
| Pumpe B (Modifikator-Lösemittel) | Acetonitril + 0,5 % Ameisensäure + 10 mM Ammoniumformiat | Interface-Temperatur | 300° C |
| Pumpe C (Modifikator-Lösemittel) | Wasser + 0,5 % Ameisensäure + 10 mM Ammoniumformiat | Heating Block | 350° C |
| Pumpe D (Make-Up-Lösemittel) | Methanol | Desolvation Line | 250° C |
| Flussrate des Make-up-Lösemittels | 0,2 mL/min |
Untersuchung der SFC-Trennbedingung bei Pestiziden
Normalerweise führt die SFC eine Gradiententrennung mit überkritischem Kohlendioxid und einem organischen Lösungsmittel (wie Methanol und Acetonitril) durch, das als Modifikator bezeichnet wird. Einige hochpolare chemische Verbindungen weisen jedoch eine starke Retention in den Säulen auf, was zu Fällen führt, in denen die Trennung und Elution mit 100 % organischem Lösungsmittel unzureichend ist. Da in diesem Experiment eine Reihe von hochpolaren Pestiziden nicht mit 100 % organischem Lösungsmittel eluiert werden konnten, wurde die Trennung durch Zugabe einer geringen Menge Wasser zum Modifikator untersucht.
Überkritisches Kohlendioxid hat eine niedrige Polarität und eine geringe Mischbarkeit mit Wasser. Dies bedeutet, dass dem Modifikator nur eine begrenzte Menge Wasser zugesetzt werden kann (normalerweise etwa 0,1 bis 10 %). Wir untersuchten daher das Trennverhalten durch Zugabe von Wasser in einer Menge, die 0,2, 4, 6, 8 und 10 % des Modifikators entspricht. Die Analyse der Signalprofile und der Trennmuster der eluierenden Komponenten zeigt bei einem Wassergehalt von 6% erste brauchbare Bedingungen. Es gab jedoch chemische Verbindungen, die auch bei dieser Bedingung nicht eluiert werden konnten.
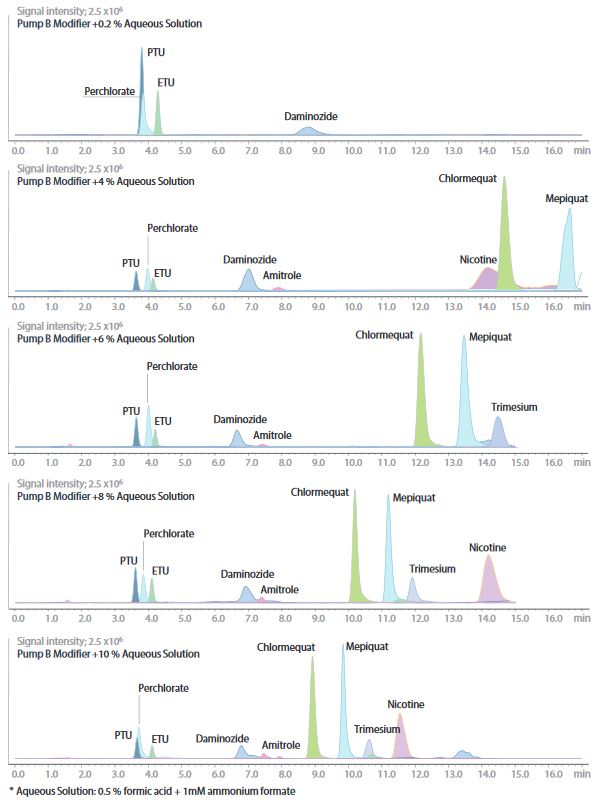
Optimierung der SFC-Trennbedingungen
Als wir die Zugabe von Wasser zum Modifikator untersuchten, konnten wir die Elution der meisten chemischen Verbindungen mit der 6 %igen wässrigen Lösung bestätigen. Nikotin und Kasugamycin, die beide eine starke Retention aufweisen, konnten jedoch nicht eluiert werden. Jede weitere Zugabe von wässriger Lösung in Gegenwart von Kohlendioxid wirkt sich negativ auf die Gradientengenauigkeit aus und kann die Stabilität der Analysemethode beeinträchtigen. Aus diesem Grund wurde die wässrige Lösung mit einer separaten Pumpe (Pumpe C) zugegeben, nachdem der Modifikator 100 % erreicht hatte (Abb. 4).
Dies ermöglichte die Elution der verbleibenden hochpolaren Pestizide und ermöglichte die Chargentrennung der hochpolaren Pestizide von logP-3,47 bis 1,96.
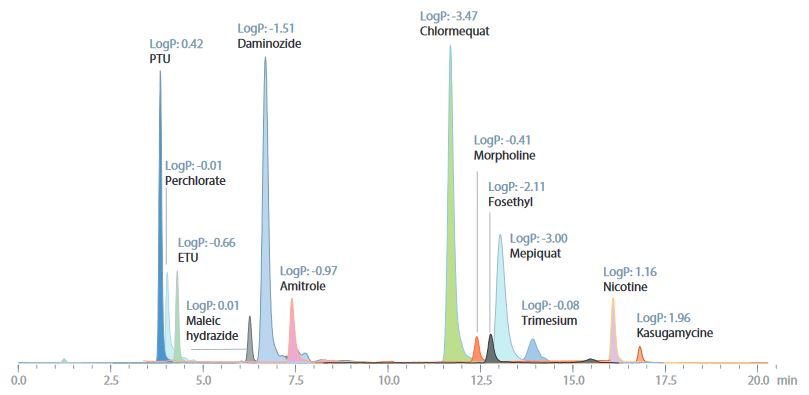
Abb. 3: MRM-Chromatogramm von hochpolaren Pestiziden unter Verwendung von SFC-MS
(Zugabe von 200 ppb Pestizid-Standardlösung zum Leinsamenextrakt unter Verwendung von QuPPe)
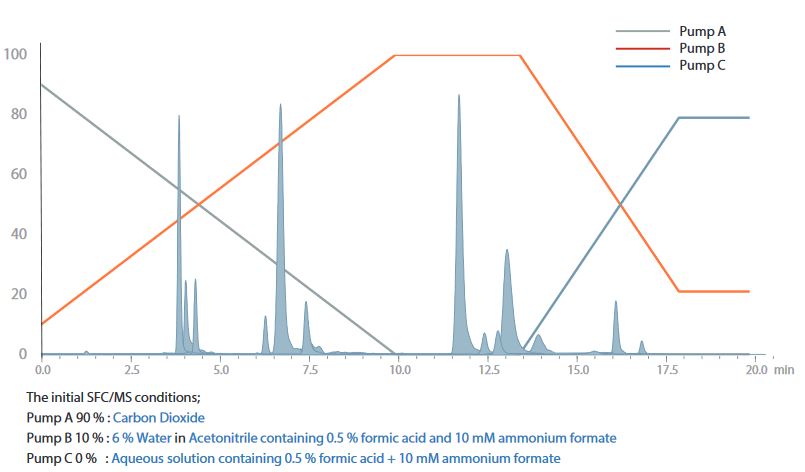
Abb. 4: Ternäres Gradienten-Programm
Probenvorbereitung und -analyse
Leinsamen und Zitrone wurden als Lebensmittelproben verwendet, und die Extraktion wurde mit einer Methode durchgeführt, die mit QuPPe übereinstimmt. (Die Extrakte wurden von Concept Life Sciences, einem analytischen Auftragslabor in Großbritannien, zur Verfügung gestellt) Zu diesen Matrixlösungen wurden Standardlösungen hochpolarer Pestizide hinzugefügt, die dann direkt in die SFC-MS/MS injiziert wurden.
Quantitative Analyse hochpolarer Pestizide
Um die quantitative Leistung der entwickelten SFC/MS-Analysemethode zu verifizieren, wurden Matrix-Kalibrierkurven mit jedem Lebensmittelextrakt erstellt, dem eine Standardlösung der hochpolaren Pestizide zugesetzt wurde. Der Bereich der Kalibrierkurven betrug 10 bis 200 ppb, und die Genauigkeit wurde mit der Methode des internen Standards bezüglich der Komponenten verifiziert, für die eine interne Standardsubstanz, die mit einem stabilen Isotop markiert war, vorhanden war.
Die für jede Probe erstellte Kalibrierkurve zeigte für alle chemischen Verbindungen unabhängig von der Probenmatrix eine günstige Linearität.
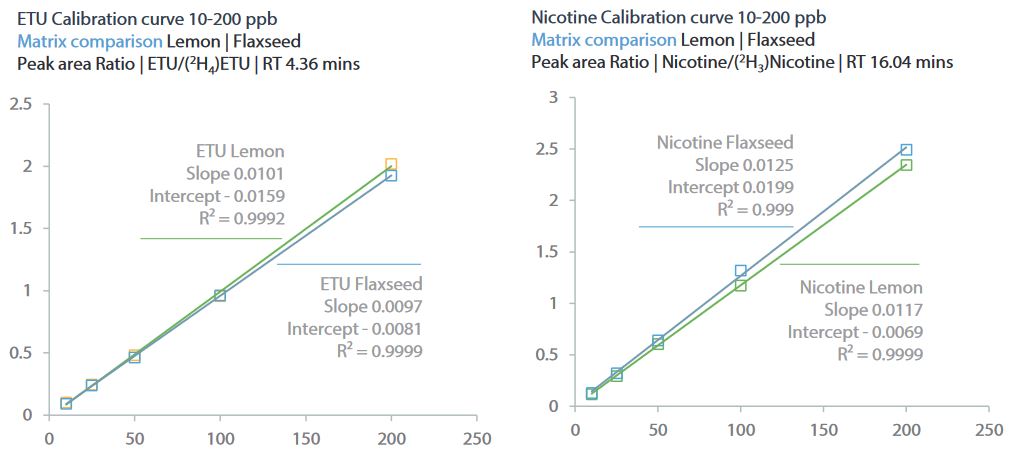
Abb. 5: Matrix-Kalibrierungskurven von repräsentativen hochpolaren Pestiziden (ETU: schnell eluierende Verbindung, Nikotin: langsam eluierende Verbindung, Proben: Zitrone, Leinsamen)
Tabelle 2: Linearität und Wiederholbarkeit der Kalibrierkurve bei 100 ppb von acht hochpolaren Pestizidkomponenten
| Verbindung | RT (min) | Interner Standard | IS RT (min) | Quan MRM | %RSD 100ppb | R2 |
|---|---|---|---|---|---|---|
| Perchlorat | 3,95 | 18O4 Perchlorat | 3,91 | 99,00>82,90 | 4,98 | 0,968 |
| ETU | 4,36 | 2H4 ETU | 4,26 | 103,10>44,05 | 4,84 | 0,999 |
| Maleinsäurehydrazid | 6,28 | 2H2 Maleinsäurehydrazid | 6,28 | 113,00>67,10 | 6,81 | 0,997 |
| Chlormequat | 11,58 | 2H4 Chlormequat | 11,54 | 121,90>58,10 | 1,75 | 1,000 |
| Fosethyl | 12,50 | 2H15 Fosethyl | 12,50 | 109,00>80,95 | 6,78 | 0,999 |
| Morpholin | 12,19 | 2H8 Morpholin | 12,23 | 87,90>70,05 | 10,74 | 0,996 |
| Mepiquat | 12,72 | 2H3 Mepiquat | 12,69 | 114,30>98,10 | 7,66 | 0,998 |
| Nikotin | 16,06 | 2H3 Nikotin | 16,03 | 163,00>130,00 | 2,31 | 0,999 |